Diagnostic Transcriptome Example
Used in Bio153H lecture using data from https://www.nature.com/articles/sdata2018136 A merged lung cancer transcriptome dataset for clinical predictive modeling. Original data was differential expression values of ~10,000 genes of ~1,000 patients with each patient being categorized as lung cancer positive or negative.
Data was preprocessed using the boruta package in r and the top 10 most important genes were selected from the total ~10,000 genes. Data was split into a training set and validation set. Various algorithms were trained and the best was tested against the confusion matrix of the validation set.
1 means cancer positive 2 means cancer negative
library(caret)## Loading required package: lattice## Loading required package: ggplot2library(lattice)
library(e1071)
library(kernlab)##
## Attaching package: 'kernlab'## The following object is masked from 'package:ggplot2':
##
## alphadataset1 <- read.csv("C:\\data\\CancerTranscriptomeTransposedImportance.csv", header = T, sep=",", colClasses = c('numeric'))
dataset1$Disease<-as.factor(dataset1$Disease)
validation_index <- createDataPartition(dataset1$Disease, p=0.80, list=FALSE)
validation <- dataset1[-validation_index,]
dataset1 <- dataset1[validation_index,]
# Run algorithms using 10-fold cross validation
control <- trainControl(method="cv", number=10)
metric <- "Accuracy"
set.seed(7)
fit.lda <- train(Disease~., data=dataset1, method="lda", metric=metric, trControl=control)
# b) nonlinear algorithms
# CART
set.seed(7)
fit.cart <- train(Disease~., data=dataset1, method="rpart", metric=metric, trControl=control)
# kNN
set.seed(7)
fit.knn <- train(Disease~., data=dataset1, method="knn", metric=metric, trControl=control)
# c) advanced algorithms
# SVM
set.seed(7)
fit.svm <- train(Disease~., data=dataset1, method="svmRadial", metric=metric, trControl=control)
# Random Forest
set.seed(7)
fit.rf <- train(Disease~., data=dataset1, method="rf", metric=metric, trControl=control)
# summarize accuracy of models
results <- resamples(list(lda=fit.lda, cart=fit.cart, knn=fit.knn, svm=fit.svm, rf=fit.rf))
# compare accuracy of models
dotplot(results)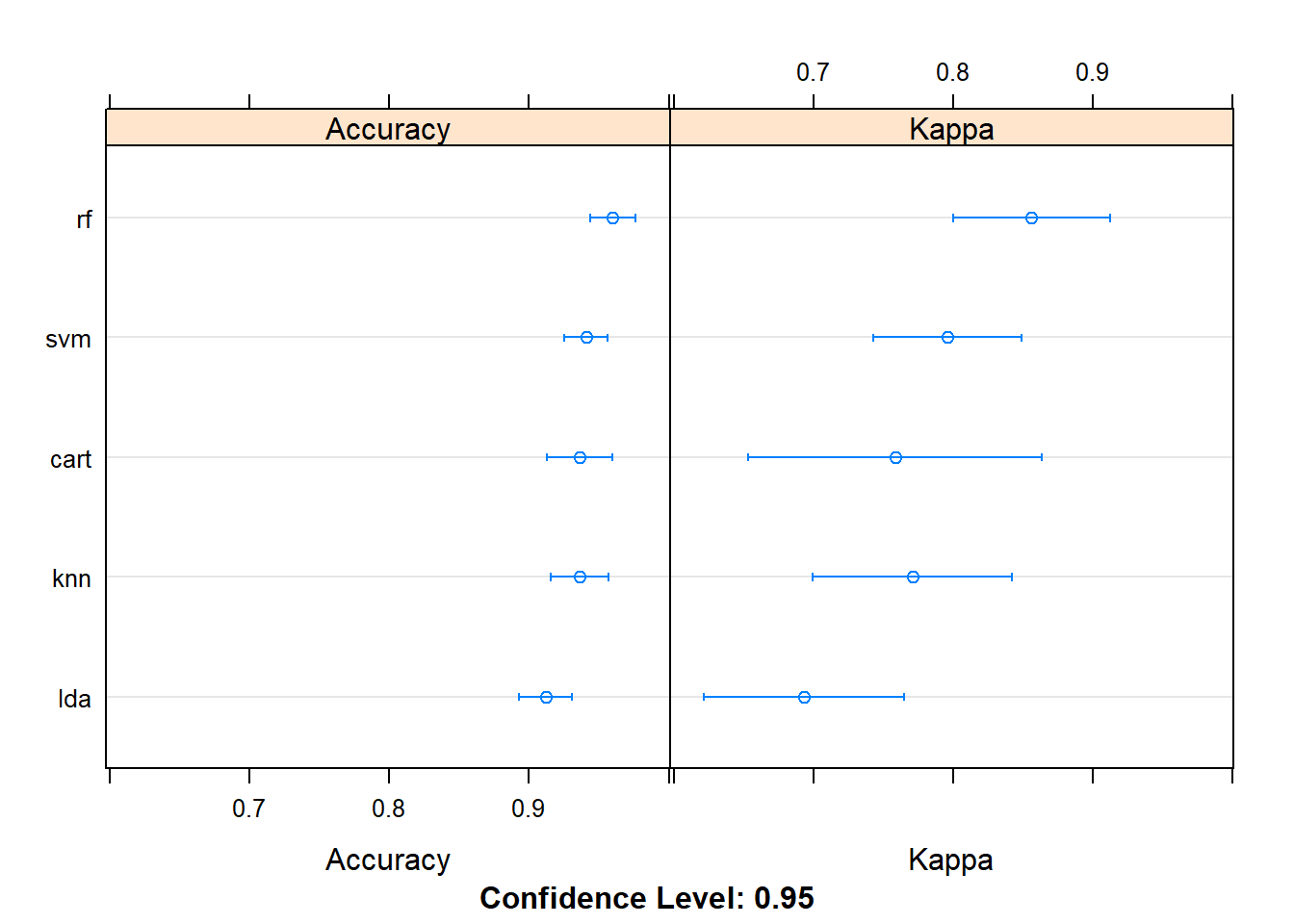
# estimate skill of LDA on the validation dataset
predictions <- predict(fit.rf, validation)
confusionMatrix(predictions, validation$Disease)## Confusion Matrix and Statistics
##
## Reference
## Prediction 1 2
## 1 184 4
## 2 0 34
##
## Accuracy : 0.982
## 95% CI : (0.9545, 0.9951)
## No Information Rate : 0.8288
## P-Value [Acc > NIR] : 1.555e-13
##
## Kappa : 0.9337
##
## Mcnemar's Test P-Value : 0.1336
##
## Sensitivity : 1.0000
## Specificity : 0.8947
## Pos Pred Value : 0.9787
## Neg Pred Value : 1.0000
## Prevalence : 0.8288
## Detection Rate : 0.8288
## Detection Prevalence : 0.8468
## Balanced Accuracy : 0.9474
##
## 'Positive' Class : 1
## 